
La evidencia actual apoya una evolución diferente, mucho más dinámica y discontinua de las placas de ateroma.
Las observaciones experimentales y humanas coinciden en que el reclutamiento de leucocitos sanguíneos mediado por la activación de las células endoteliales que recubren el lumen arterial es un fenómeno temprano en la formación de la lesión.
Dr. Alfonso Galán González – Equipo Médico Neolife
Siguiendo con estos artículos sobre ateroesclerosis vamos a hablar en esta nueva entrega sobre cómo progresan las lesiones ateroescleróticas, las placas.
Conocer cómo se forman y qué factores influyen en su evolución es vital para evitar su progresión.
LDL oxidado e inicio de lesiones
La mayoría de las revisiones de los mecanismos de la aterosclerosis postulan un papel fundamental para el LDL oxidado como el motor principal de esta enfermedad. Ya hace dos años escribimos sobre él en estas páginas (aquí). Pero, a pesar de un gran cuerpo de evidencia en este sentido en estudios animales, los estudios humanos que confirmen su papel causal no son tantos. Los ensayos realizados con vitaminas antioxidantes o con un muy efectivo antioxidante lipófilo no han reducido los eventos ateroscleróticos. Quizá la respuesta esté en la observación de que cuando los lípidos oxidados se unen al plasminógeno, pueden activar la fibrinólisis. Por lo tanto, los lípidos oxidados pueden promover la aterogénesis pero también estimular la trombólisis, un efecto opuesto que podría contribuir a esta falta de beneficio neto en los ensayos de estrategias antioxidantes.
Por tanto, es inteligente que busquemos explicaciones más allá de la hipótesis de oxidación para comprender cómo el LDL causa aterosclerosis.
El LDL que se agrega en la íntima en asociación con proteoglicanos o respuestas inmunes adaptativas a las LDL nativas, proporcionan mecanismos alternativos a través de los cuales esta lipoproteína promueve la aterogénesis.
Independientemente del desencadenante o desencadenantes iniciales, las observaciones experimentales y humanas coinciden en que el reclutamiento de leucocitos sanguíneos mediado por la activación de las células endoteliales que recubren el lumen arterial es un fenómeno temprano en la formación de la lesión.
El endotelio en reposo resiste la unión de los leucocitos sanguíneos. Sin embargo, en un ambiente aterogénico, las células endoteliales pueden expresar moléculas de adhesión leucocitaria que median la adhesión de los glóbulos blancos a la superficie íntima. Mediadores químicos dirigen la migración de los leucocitos adherentes hacia la íntima arterial. Los fagocitos mononucleares proliferan dentro de la capa íntima (el sitio de inicio de la lesión). Estas células engullen los lípidos y se convierten en células espumosas, el sello distintivo de las lesiones ateroscleróticas. Los linfocitos T, los protagonistas de la respuesta inmune adaptativa, interactúan con las células de la inmunidad innata dentro de la íntima.
La cooperación entre estos constituyentes celulares de la inmunidad innata y adaptativa estimula la producción de citoquinas proinflamatorias que mantienen y amplifican la respuesta inflamatoria local.
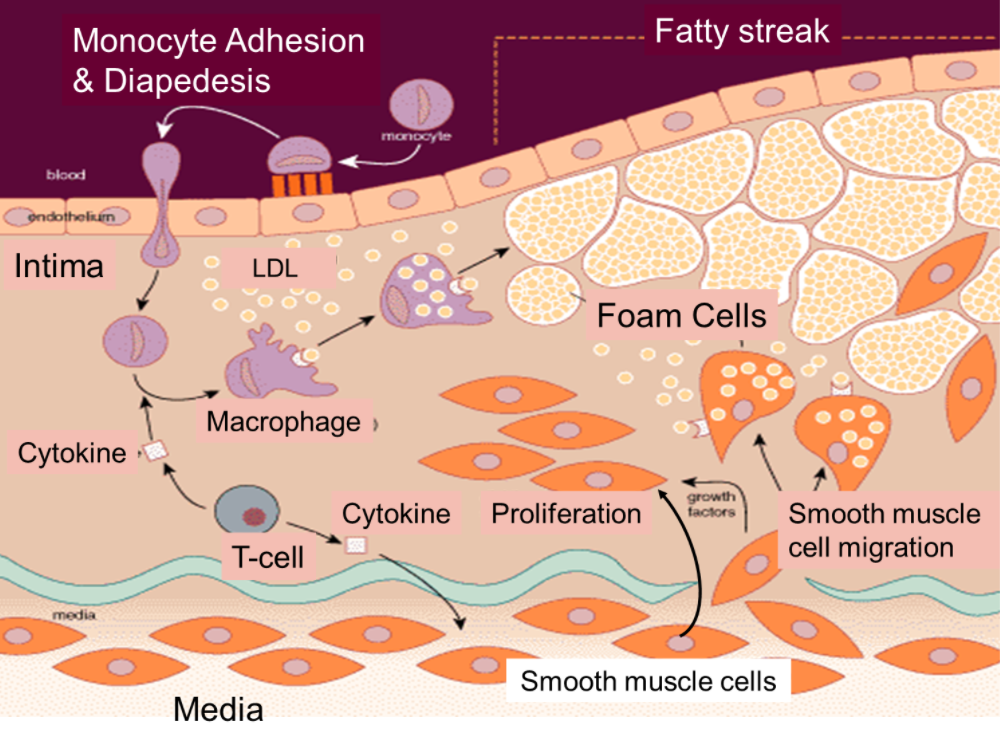
En los seres humanos la íntima contiene células de músculo liso residentes. Otras células de músculo liso (que generalmente se encuentran en la media) pueden penetrar en la íntima, donde se unen a las células de músculo liso residentes para promover la acumulación de matriz extracelular que estas células sintetizan dentro de esta capa íntima en expansión.
Inexorabilidad de la progresión del ateroma
Muchos han considerado la aterosclerosis como un proceso “degenerativo” inevitable que progresa continuamente con el tiempo, pero la evidencia actual apoya una evolución mucho más dinámica y discontinua de las placas de ateroma. Los episodios de inflamación sistémica o inflamación regional alejados de la propia placa aterosclerótica pueden provocar “crisis” en la evolución de la placa y estimular una ronda de activación inflamatoria que puede promover la migración celular, la proliferación, la progresión de la lesión y su complicación.
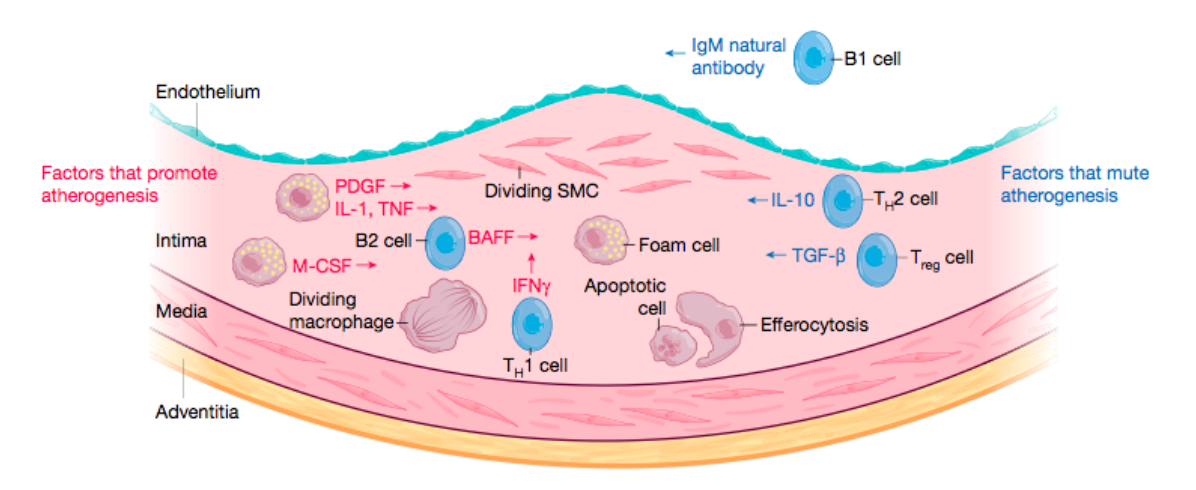
Las células del músculo liso arterial, que normalmente residen en la capa media de la arteria, entran en la capa íntima, donde pueden proliferar y sufrir metaplasia para convertirse en células parecidas a macrófagos.
Es posible que la aterosclerosis no se desarrolle de forma continua, sino de una forma en que se alternan fases de relativa inactividad con períodos de rápido crecimiento.
Cada vez más evidencia emergente apunta a la hematopoyesis (formación de células sanguíneas) como un contribuyente clave a la evolución de las lesiones y como nexo entre la inflamación regional, los estímulos ambientales y la aterogénesis.
Estrés mental, alteraciones del sueño y lesiones o infecciones en otro punto de nuestro cuerpo puede estimular la hematopoyesis en la médula ósea, aprovisionando de leucocitos que pueden poblar la placa. También la hematopoyesis extramedular, así como la movilización de grupos ya formados de leucocitos en el bazo, proporcionan más leucocitos que pueden anidar en nuestras placas de ateroma en situaciones de estrés.
De hecho, el trabajo que identificó CHIP (explicado en el artículo previo de esta serie sobre ateroesclerosis) como factor de riesgo subraya el vínculo entre aterosclerosis y hematopoyesis. Estas observaciones han abierto una ventana a la patogénesis de la aterosclerosis y proporcionar un vínculo entre la oncogénesis y la aterogénesis que era insospechada sólo hace unos pocos años.
La muerte de fagocitos mononucleares en la lesión, y su aclaramiento ineficaz (eferocitosis defectuosa), promueve la formación del núcleo lipídico o necrótico de la lesión aterosclerótica.
La progresión de la lesión puede ocurrir silenciosamente durante muchas décadas. De hecho, a muchos individuos jóvenes o de mediana edad cuando les hacemos pruebas de imagen tienen lesiones ateroscleróticas subclínicas.
“Placas vulnerables”
Los episodios agudos como los infartos de miocardio y los accidentes cerebrovasculares isquémicos (Ictus) que complican la aterosclerosis surgen de la trombosis o formación de coágulos sanguíneos; una alteración física de las placas ateroscleróticas provoca la mayoría de las trombosis agudas.
El concepto de “placa vulnerable” ha recibido una considerable atención6. Una fractura de la capa fibrosa de la placa (que recubre el núcleo necrótico) expone la sangre circulante y sus proteínas de coagulación a sustancias trombogénicas dentro de la placa, desencadenando una trombosis aguda.
El casquete fibroso debe su resistencia a la tracción en gran parte al colágeno intersticial. El adelgazamiento de la capa fibrosa surge de una disminución de la síntesis de colágeno y un aumento de su degradación asociado a la inflamación y a la sobreexpresión de colagenasas por las células inflamatorias. Los estudios de autopsias han implicado la rotura del casquete fibroso como la causa de la mayoría de los síndromes coronarios agudos mortales.
Pero estos estudios post-mortem no suelen mirar cuántas de estas placas con estas características NO están causando complicaciones trombóticas agudas.
La evidencia reciente ha proporcionado esta información que faltaba y ha demostrado que las placas cubiertas de esta capa fina rara vez causan eventos clínicos, por lo que quizá el término “placa vulnerable” no es el más adecuado.
En una era de intensa reducción de lípidos, las placas de la clásica morfología vulnerable están en declive. Otro mecanismo de alteración de la placa conocido como erosión superficial, parece estar en aumento y probablemente tiene una fisiopatología diferente. En este caso, el fenómeno desencadenante de una obstrucción coronaria no implica fisura o rotura del casquete fibroso de la placa, sino más bien una discontinuidad en el revestimiento endotelial de la íntima. La aplicación de una modalidad de imagen intravascular conocida como tomografía de coherencia óptica permite identificar la rotura de la placa y ha llevado al desarrollo de criterios para el diagnóstico de erosión probable o definitiva en individuos con síndromes coronarios agudos. Los mecanismos de erosión involucran lesión endotelial, la participación de leucocitos polimorfonucleares y trampas extracelulares de neutrófilos (NET –ver más abajo-) como contribuyentes locales a la formación y propagación de trombos.
Resumiendo y representando gráficamente estas dos formas de complicación de la placa para facilitar la comprensión:
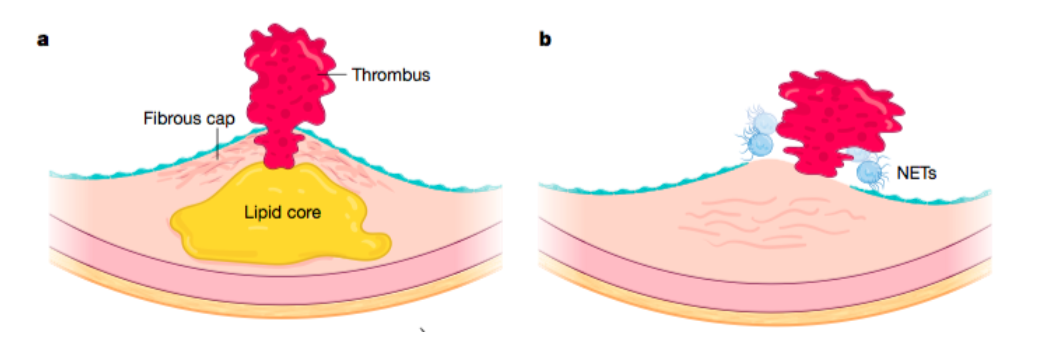
En a, vemos la Rotura de placa. Esto implica una fractura o fisura del casquete fibroso que recubre el núcleo lipídico de la placa. Esta alteración física permite el contacto de los factores de coagulación de la sangre con material trombogénico (principalmente el potente factor tisular procoagulante) dentro de la placa. La trombosis resultante puede obstruir el flujo sanguíneo y provocar isquemia cardíaca. Este mecanismo explica aproximadamente dos tercios de los infartos agudos de miocardio, pero parece estar disminuyendo; las terapias preventivas actuales conducen a una reducción de la acumulación de lípidos dentro de las placas y al refuerzo de la capa fibrosa.
Y en b, observamos lo que sería la erosión superficial. Esta causa de formación de coágulos en las arterias coronarias implica una suerte de descamación de la monocapa endotelial. Los granulocitos atrapados en la placa o adheridos a la membrana basal de la íntima pueden formar lo que se ha venido a llamar trampas extracelulares de neutrófilos (NET). Los NET son una mezcla de hebras de ADN nuclear que se han desenrollado, varias proteínas granulares de neutrófilos y otras proteínas que se unen de la sangre, formando una especie de reactivo sólido en la superficie de la íntima que puede propagar la inflamación y la trombosis.
En la próxima (y última entrega) de estos artículos sobre las evidencias actuales sobre ateroesclerosis trataremos de poner todo esto en conjunto para darle sentido desde el punto de vista clínico.
BIBLIOGRAFÍA
(1) Leibundgut, G. et al. Oxidized phospholipids are present on plasminogen, affect fibrinolysis, and increase following acute myocardial infarction. J. Am. Coll. Cardiol. 59, 1426–1437 (2012).
(2) Kubo, T. et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J. Am. Coll. Cardiol. 55, 1590–1597 (2010).
(3) Vergallo, R. & Crea, F. Atherosclerotic plaque healing. N. Engl. J. Med. 383, 846–857 (2020).
(4) Schloss, M. J., Swirski, F. K. & Nahrendorf, M. Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ. Res. 126, 1242–1259 (2020).
(5) Tuzcu, E. M. et al. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circulation 103, 2705–2710 (2001).
(6) Waksman, R. et al. The lipid-rich plaque study of vulnerable plaques and vulnerable patients: study design and rationale. Am. Heart J. 192, 98–104 (2017).
(7) Libby, P. & Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur. Heart J. 36,2984–2987 (2015).
(8) Arbab-Zadeh, A. & Fuster, V. The myth of the “vulnerable plaque”: transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J. Am. Coll. Cardiol. 65, 846–855 (2015).
(9) Franck, G. et al. Haemodynamic stress-induced breaches of the arterial intima trigger inflammation and drive atherogenesis. Eur. Heart J. 40, 928–937 (2019).
