
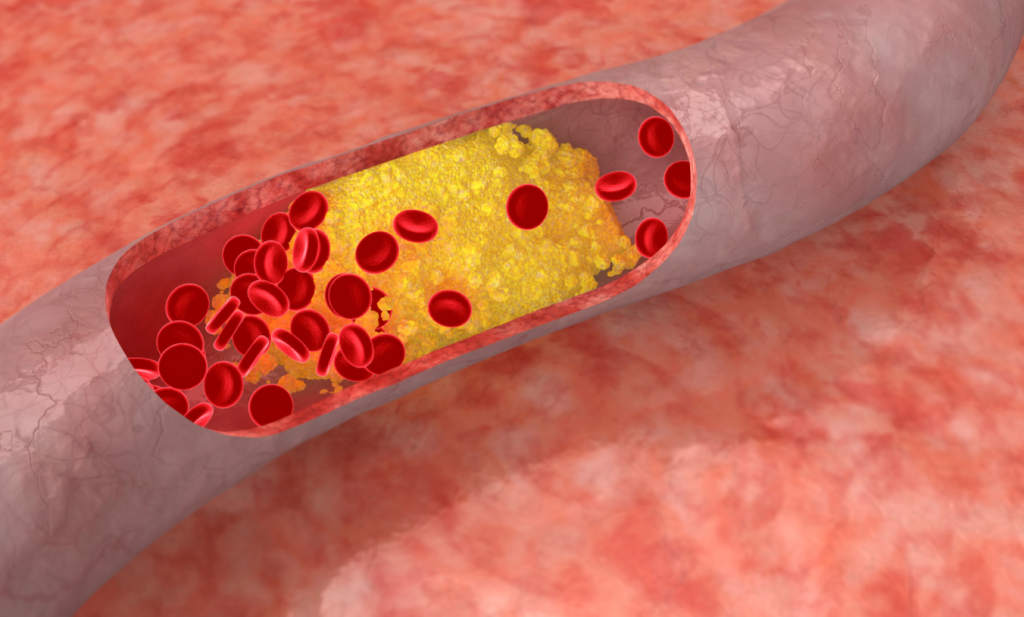

Current evidence supports a different, much more dynamic and discontinuous evolution of atheroma plaques.
Experimental and human observations support that recruitment of blood leukocytes mediated by activation of endothelial cells lining the arterial lumen is an early phenomenon in lesion formation.
Dr. Alfonso Galán González – Neolife Medical Team
Continuing with these articles on atherosclerosis, in this new installment, we will delve into how atherosclerotic lesions, plaques, are formed.
Knowing how they are formed and what factors influence their evolution is essential to preventing their progression.
Oxidized LDL and onset of lesions
Most reviews of the mechanisms of atherosclerosis postulate a pivotal role for oxidized LDL as the main driver of this disease. Two years ago, we wrote about this (here). But, despite a large body of evidence in this regard in animal studies, human studies confirming its causal role are not as numerous. Trials with antioxidant vitamins or a very effective lipophilic antioxidant have not reduced atherosclerotic events. Perhaps the answer lies in the observation that when oxidized lipids bind to plasminogen, they can activate fibrinolysis.. This means that oxidized lipids may promote atherogenesis, but also stimulate thrombolysis, an opposite effect that may contribute to this lack of net benefit in trials of antioxidant strategies.
Therefore, it is smart for us to look beyond the oxidation hypothesis for explanations of how LDL causes atherosclerosis.
LDL aggregating in the intima, in association with proteoglycans or adaptive immune responses to native LDL, provide alternative mechanisms through which this lipoprotein promotes atherogenesis.
Regardless of the initial trigger(s), experimental and human observations support that recruitment of blood leukocytes mediated by activation of endothelial cells lining the arterial lumen is an early phenomenon in lesion formation.
The resting endothelium resists the binding of blood leukocytes. However, in an atherogenic environment, endothelial cells can express leukocyte adhesion molecules that mediate the adhesion of white blood cells to the intimal surface. Chemical mediators direct the migration of adherent leukocytes into the arterial intima. Mononuclear phagocytes proliferate within the intimal layer (the site where the injury begins). These cells engulf lipids and become foam cells, the hallmark of atherosclerotic lesions. T-cells, the protagonists of the adaptive immune response, interact with cells of innate immunity within the intima.
Cooperation between these cellular constituents of innate and adaptive immunity stimulates the production of proinflammatory cytokines that maintain and amplify the local inflammatory response.
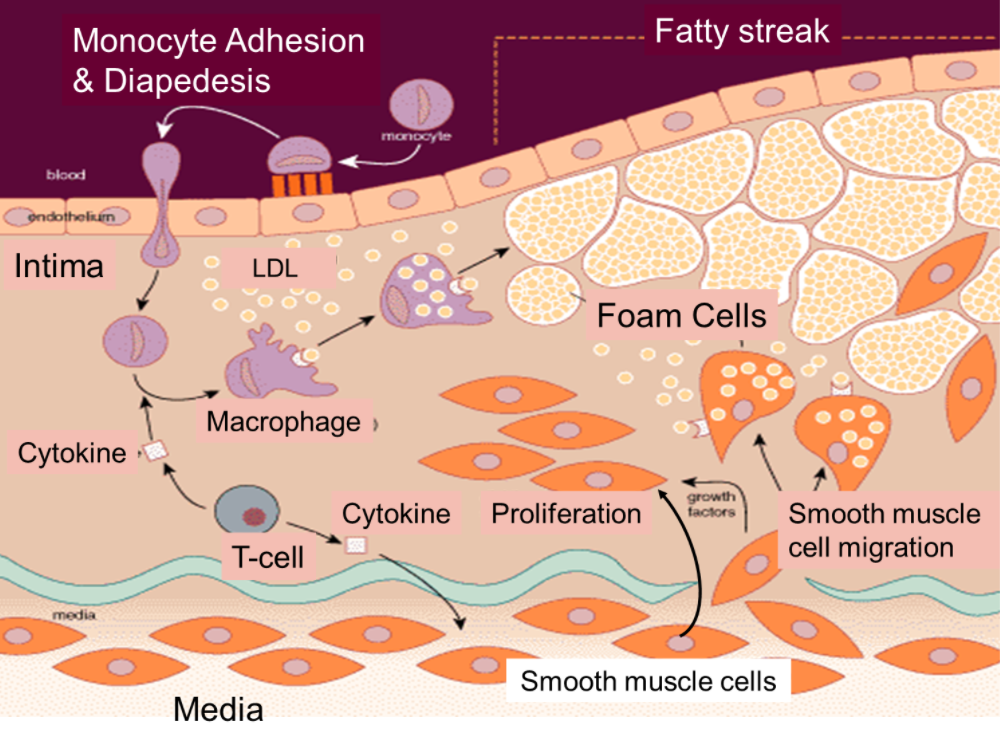
In humans, the intima contains resident smooth muscle cells. Other smooth muscle cells (usually found in the media) can penetrate into the intima, where they join the resident smooth muscle cells to promote the accumulation of extracellular matrix that these cells synthesize within this expanding intimal layer.
Inexorability of atheroma progression
Many have regarded atherosclerosis as an inevitable “degenerative” process that progresses continuously over time, but current evidence supports a much more dynamic and discontinuous evolution of atherosclerotic plaques. Episodes of systemic inflammation or regional inflammation away from the atherosclerotic plaque itself may provoke “crises” in plaque evolution and stimulate a round of inflammatory activation that may lead to cell migration, proliferation, lesion progression, and complication.
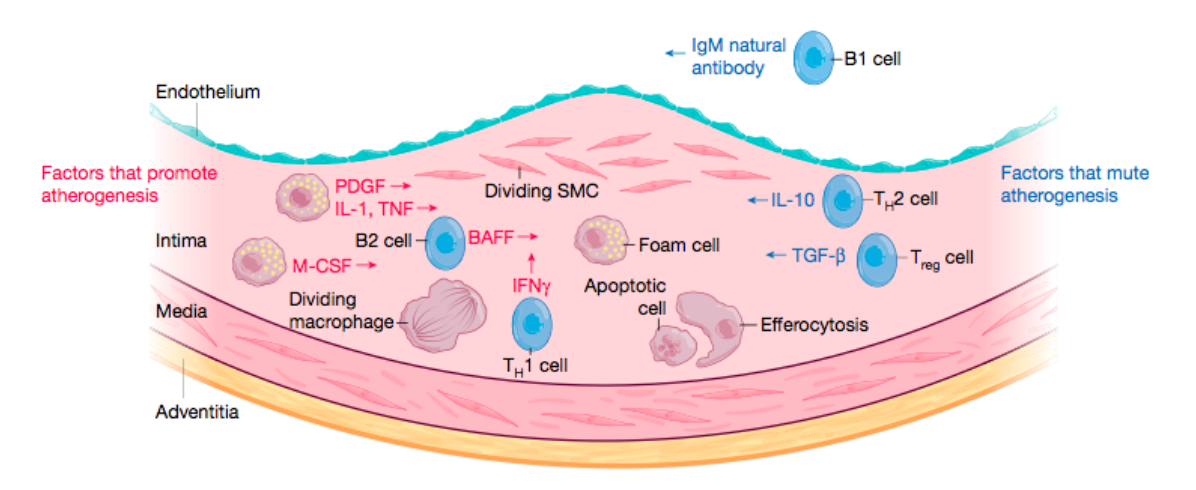
Arterial smooth muscle cells, which normally reside in the media layer of the artery, enter the intimal layer, where they can proliferate and undergo metaplasia to become macrophage-like cells.
Atherosclerosis may not develop continuously, but rather in a way that alternates phases of relative inactivity with periods of rapid growth.
Increasingly emerging evidence points to hematopoiesis (blood cell formation) as a key contributor to lesion evolution and as a link between regional inflammation, environmental stimuli, and atherogenesis..
Mental stress, sleep disturbances, and injuries, or infections elsewhere in our body can stimulate hematopoiesis in bone marrow, supplying white blood cells that can populate the plaque. Also, extramedullary hematopoiesis, as well as the mobilization of already formed pools of white blood cells in the spleen, provide more white blood cells that can nest in our atheromatous plaques under stressful situations.
In fact, the work that identified clonal hematopoiesis of indeterminate potential (also known as CHIP and explained in the previous article in this series on atherosclerosis) as a risk factor underscores the link between atherosclerosis and hematopoiesis. These observations have opened a window into the pathogenesis of atherosclerosis and provide a link between oncogenesis and atherogenesis that was unsuspected only a few years ago.
The death of mononuclear phagocytes in the lesion, and their ineffective clearance (defective efferocytosis), promotes the formation of the lipid or necrotic core of the atherosclerotic lesion.
Lesion progression may occur silently over many decades. In fact, many young or middle-aged individuals have subclinical atherosclerotic lesions when we perform imaging tests..
“Vulnerable plaques”
Acute episodes such as myocardial infarctions and ischemic strokes (Ictus) that complicate atherosclerosis arise from thrombosis or blood clot formation; a physical disruption of atherosclerotic plaques causes the most acute thromboses.
The concept of “vulnerable plaque” has received considerable attention6. A fracture of the fibrous cap of the plaque (overlying the necrotic core) exposes the circulating blood and its clotting proteins to thrombogenic substances within the plaque, triggering acute thrombosis.
The fibrous cap owes its tensile strength largely to interstitial collagen. Thinning of the fibrous cap arises from a decrease in collagen synthesis and an increase in its degradation associated with inflammation and overexpression of collagenases by inflammatory cells. Autopsy studies have implicated fibrous cap rupture as the cause of most fatal acute coronary syndromes.
But these post-mortem studies don’t usually look at how many of these plaques with these characteristics are NOT causing acute thrombotic complications.
Recent evidence has provided this missing information and has shown that plaques covered with this thin layer rarely cause clinical events, so perhaps the term “vulnerable plaque” is not the most appropriate term.
In an era of intense lipid depletion, plaques of classic vulnerable morphology are in decline. Another mechanism of plaque disruption known as surface erosion appears to be on the rise and probably has a different pathophysiology. In this case, the triggering phenomenon of coronary obstruction does not involve fissure or rupture of the fibrous cap of the plaque, but rather a discontinuity in the endothelial lining of the intima. The application of an intravascular imaging technique known as optical coherence tomography allows the identification of plaque rupture and has led to the development of criteria for the diagnosis of probable or definite erosion in individuals with acute coronary syndromes. Erosion mechanisms involve endothelial injury, the participation of polymorphonuclear white blood cells, and neutrophil extracellular traps (NETs –see below–) as local contributors to thrombus formation and propagation..
To facilitate understanding, let’s summarize and graphically represent these two complications we may see in the plaque:
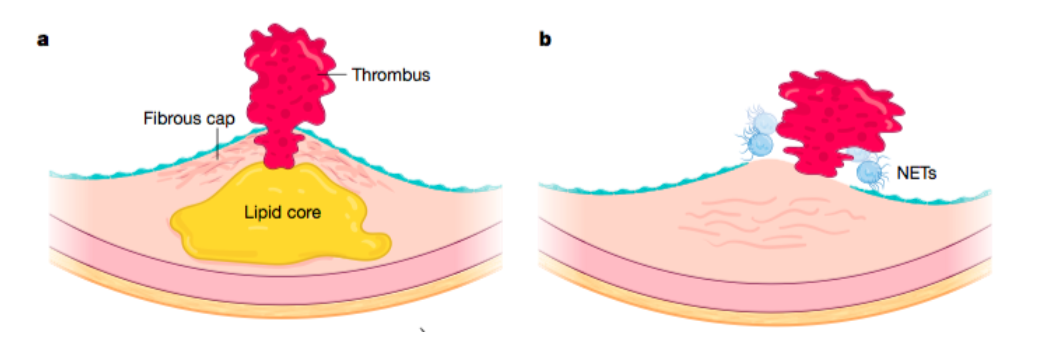
In a, we see plaque rupture. This involves a fracture or fissure of the fibrous cap overlying the lipid core of the plaque. This physical alteration allows contact of blood clotting factors with thrombogenic material (mainly the potent procoagulant tissue factor) within the plaque. The resulting thrombosis can obstruct blood flow and cause cardiac ischemia. This mechanism explains approximately two-thirds of acute myocardial infarctions, but appears to be declining; current preventive therapies lead to a reduction in lipid accumulation within plaques and reinforcement of the fibrous cap.
And in b, we observe what would be surface erosion. This cause of clot formation in the coronary arteries involves a kind of endothelial monolayer desquamation. Granulocytes trapped in the plaque or attached to the basement membrane of the intima may form neutrophil extracellular traps (NETs). NETs are a combination of nuclear DNA strands that have unwound, various neutrophil granular proteins and other proteins that bind from the blood, forming a kind of solid reactant on the surface of the intima that can spread inflammation and thrombosis.
In the next (and final) installment of these articles on the current evidence on atherosclerosis, we will try to put all this together to make sense of it from a clinical point of view.
BIBLIOGRAPHY
(1) Leibundgut, G. et al. Oxidized phospholipids are present on plasminogen, affect fibrinolysis, and increase following acute myocardial infarction. J. Am. Coll. Cardiol. 59, 1426–1437 (2012).
(2) Kubo, T. et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J. Am. Coll. Cardiol. 55, 1590–1597 (2010).
(3) Vergallo, R. & Crea, F. Atherosclerotic plaque healing. N. Engl. J. Med. 383, 846–857 (2020).
(4) Schloss, M. J., Swirski, F. K. & Nahrendorf, M. Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ. Res. 126, 1242–1259 (2020).
(5) Tuzcu, E. M. et al. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circulation 103, 2705–2710 (2001).
(6) Waksman, R. et al. The lipid-rich plaque study of vulnerable plaques and vulnerable patients: study design and rationale. Am. Heart J. 192, 98–104 (2017).
(7) Libby, P. & Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur. Heart J. 36,2984–2987 (2015).
(8) Arbab-Zadeh, A. & Fuster, V. The myth of the “vulnerable plaque”: transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J. Am. Coll. Cardiol. 65, 846–855 (2015).
(9) Franck, G. et al. Haemodynamic stress-induced breaches of the arterial intima trigger inflammation and drive atherogenesis. Eur. Heart J. 40, 928–937 (2019).