

Despite its fundamental role, the mitochondrion has not been getting the attention it deserves when it comes to COVID-19. In this blog post, we’ll explain the part it plays in this disease.
Patients with COVID-19 mainly present a lower respiratory tract infection. Unfortunately, a significant number of patients develop what is called a “cytokine storm”, a hyper-inflammatory state associated with oxidative stress, deregulation of iron homeostasis, hypercoagulability, and clot formation.
Dr. Alfonso Galán González – Neolife Medical Team
The fundamental role that mitochondria play in platelet function and survival
In this new blog post, we wish to present the latest research, which links mitochondrial dysfunction and alterations of the gut microbiota with the pathogenesis of coronavirus disease; in other words, they are intimately involved in the way COVID-19 affects us and causes us harm.
In another blog post, we discussed how mitochondria are involved in the aging process (here) and gut microbiota and the causes and consequences of dysbiosis.
Some evidence, which we have already discussed here, was already pointing to the mitochondria and their energy production mechanisms as something that was affected in one way or another in COVID-19 disease. In this blog post, and as simply as possible, we will break down what we know about mitochondrial involvement and our gut flora in coronavirus disease, and how mitochondrial dysfunction in the disease may also affect platelet survival and apoptosis, and potentially increase the risk of clot formation.
A brief introduction
It is common knowledge that SARS-CoV-2 is a betacoronavirus that emerged in Wuhan, China in December 2019. Since then, the pandemic has spread rapidly, causing over 2.7 million deaths worldwide at the time of writing.
Essentially, patients with COVID-19 present a lower respiratory tract infection. Unfortunately, a significant number of patients develop what is called a “cytokine storm”, a hyper-inflammatory state associated with oxidative stress, deregulation of iron homeostasis, hypercoagulability, and clot formation.
During this time, several biomarkers have been associated with a worse prognosis and mortality in critically ill patients, mainly lymphopenia, increased D-dimer, and IL-6. A prospective study of large numbers of ICU patients reported a higher risk of death for every 10% increase in IL-6 or D-dimer, pointing to the close link between COVID-19 disease and systemic inflammation and endothelial damage.
Higher levels of Ferritin are also a predictor of mortality, as is oxidative stress. A great deal of evidence links inflammation with oxidative stress.
Despite its fundamental role in controlling oxidation and the formation of reactive oxygen species (ROS), not much has been said about mitochondria as an important part of COVID-19.
Next, we will explain the theoretical role of inflammatory signals when it comes to aggravating oxidative mitochondrial damage and how this contributes to alterations in coagulation, ferroptosis (cell death dependent on iron), and dysbiosis. We will also see how extracellular mitochondria, in particular platelet mitochondria, affect coagulation and clot formation.
Mitochondria, oxidative stress, and inflammation
Normally, our tissues require a large number of functioning mitochondria to provide energy and regulate cellular functions based on our needs. If there is a higher demand, we create more mitochondria through a process called mitogenesis, while excess mitochondria are eliminated through a process called mitophagy. Mitochondrial defects have been implicated in many pathologies, like diabetes, cardiovascular disease, gastrointestinal diseases, cancer, and the aging process. Mitochondria are the biggest source of reactive oxygen species (ROS), which on the one hand leads to normal cellular functioning, but on the other hand, has been associated with an increase in intracellular oxidative stress.
Pro-inflammatory cytokines induce an increase in mitochondrial ROS. These inflammatory cytokines, like TNF-alpha and IL-6, are found in the blood of COVID patients, affecting mitochondrial oxidative phosphorylation, which leads to the production of ATP (our energy currency) and to the formation of ROS. This affects mitochondria in many ways leading to cell death (apoptosis) in the worst case scenario. Additionally, when these damaged mitochondria release their contents into the cell’s cytosol or extracellular space, this leads to the production of pro-inflammatory cytokines, like IL-1 or IL-6. Both are markers in severe cases of COVID-19.
Many studies have also shown the impact of mitochondrial dysfunction on immune response. For example, the production of pro-inflammatory cytokines is greatly increased in lung cells with dysfunctional mitochondria.
Iron and mitochondrial dysfunction
Studies of markers in COVID-19 have shown without any doubt that excess ferritin is associated with the severity of the disease and a worse prognosis. Ferritin, in addition to being a marker of inflammation, is released by the cells that die.
One of the iron-mediated oxidative stress targets is none other than the mitochondria. The function of the mitochondria depends on the right amount of iron, and an alteration in its iron levels will alter its function, leading to stress and cell death.
Moreover, we know that non-protein circulating iron catalyzes the formation of higher levels of ROS that cause damage to cellular and mitochondrial components, as well.
Ferroptosis is a recently discovered phenomenon of programmed cell death associated with iron accumulation. It has been shown in numerous infections by different microorganisms. Peculiarly, ferroptosis causes irreversible alterations in mitochondrial morphology
.
This data provides a solid basis for the idea that mitochondrial dysfunction plays a key role in COVID-19 disease.
Thrombopenia and hypercoagulability in COVID
It is clear at this point that coagulation abnormalities are closely associated with COVID mortality. The comparison of coagulation parameters between survivors and non-survivors indicates a higher D-dimer, higher fibrin degradation products, higher prothrombin time, and higher activated partial thromboplastin.
Also, a decrease in the number of platelets has been associated with an increase in mortality and is more common in more severe cases especially associated with disseminated intravascular coagulation (DIC)
Extracellular mitochondria and platelet mitochondria
We may curiously find mitochondria outside the cells; this is something that is not well known. Extracellular mitochondria may be found cell-free, enclosed by a membrane such as inside platelets, or in vesicles. Out-of-cell mitochondria may induce paracrine or endocrine responses and regulate cell-to-cell communication, regeneration, hazard detection, and cause immune responses. In addition to extracellular mitochondria, the release of mitochondrial DNA (MDNA) has been described during apoptosis.
The potential role of cell-free circulating mitochondria or their blood derivatives in patients with COVID-19 has not yet been determined, but it is something that may have therapeutic implications, especially with the use of plasma obtained from recovered patients to treat sick patients. But let’s delve a little deeper.
A platelet is a small anucleate blood cell with the primary physiological and pathological function of hemostasis and wound healing. In the absence of an all-controlling nucleus, platelet health is largely determined by the health of its mitochondria.
The role that mitochondria play in platelet function and survival is fundamental. Mitochondrial dysfunction in the disease may also affect platelet survival and apoptosis, and potentially increase the risk of clot formation. More importantly, it has recently been shown that apoptotic platelets may induce clotting e”50 times faster than normal platelets.
An increase in mitochondrial ROS production in platelets leads to severe oxidative stress that alters ATP production and mitochondrial membrane potential, leading to increased platelet activation.
This may explain the small number of platelets in COVID-19 disease despite the onset of thrombosis. Additionally, patients with COVID-19 are likely to suffer from impaired mitophagy due to the stress caused by hyper-inflammation. In a healthy context, mitophagy protects platelets from oxidative stress and mitochondrial destruction by eliminating damaged mitochondria to prevent platelet apoptosis; when platelet mitophagy, which is involved in COVID-19 pathogenesis, is altered, there is an increase in platelet apoptosis that contributes to an increase in thrombosis.
Therefore, preserving the mitochondrial function of platelets may be an additional means of decreasing the risk of potentially fatal thrombotic events in COVID-19.
On the other hand, by linking this to the previous point of iron overload in COVID, evidence suggests that iron overload is an agent of platelet dysfunction. Excess iron alters mitochondrial function and promotes oxidative stress. It is believed that there is a link between iron overload and elevated ROS.
Moreover, activated platelets may release mitochondria into microvesicles into the extracellular medium when exposed to oxidative stress. These mitochondria may be lysed by generating inflammatory mediators that promote endothelial inflammation.
Therefore, extracellular mitochondria and their “derivatives” may represent critical mediators in the progression of the inflammatory environment that leads to coagulopathy associated with inflammatory response pathways.
Cell-free mitochondria and mitochondria in microvesicles are a major subset of extracellular vesicles released by activated monocytes, and their pro-inflammatory activity in endothelial cells is determined by the activation status of their parent cells. Therefore, mitochondria may be critical intercellular mediators in cardiovascular disease and other inflammatory environments.
COVID and dysbiosis
It is very important to consider the link between mitochondria and microbiota in COVID-19 for several reasons:
- First, some patients have concurrent gastrointestinal symptoms, including diarrhea.
- The virus’ genetic material has been detected in the feces of patients with COVID-19.
These observations indicate the ability of SARS-CoV-2 to colonize the gastrointestinal tract, which would disrupt the gut microbiota. Interestingly, fecal metabolomic analysis suggested possible pathways linking gut microbiota to inflammation, which would explain the predisposition of certain individuals to develop severe COVID-19. Addressing the impact of the COVID-19 microbiota on mitochondrial function would provide new avenues for therapeutic strategies.
The interaction between the microbiota and the mitochondria appears to occur both ways through endocrine, immune, and humoral links
.
The gut microbiota influences mitochondrial functions related to energy production, mitochondrial biogenesis, redox balance, and inflammatory cascades. On the other hand, mitochondrial functions may alter the composition and activity of the gut microbiota. In fact, as described above, under stressful conditions such as bacterial or viral infections, mitochondria may modulate immune responses that lead to increased inflammation. This unbalanced immune response may result in dysbiosis.
Conclusions
Let’s try to sum this all up in a few words and present some takeaways:
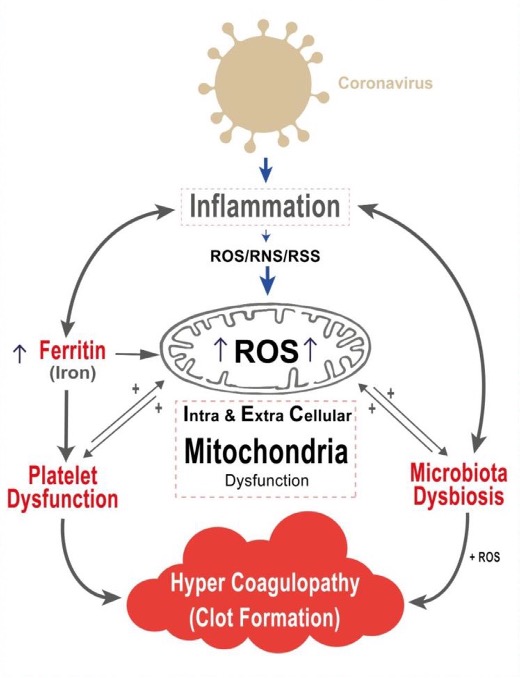
- COVID-19 disease involves an inflammatory state.
- This inflammatory state involves the production of large numbers of inflammatory cytokines and is linked to the presence of hyperferritinemia (iron overload), leading to higher oxidative stress and cellular damage.
- The mitochondrion is the fundamental organelle in the generation of reactive oxygen species (ROS).
- Higher levels of ROS lead to intra and extracellular mitochondrial damage.
- This mitochondrial damage leads to microbiota dysbiosis.
- Additionally, platelet dysfunction plays a fundamental role in clot formation.
- Mitochondrial damage leads to the release of its contents, which aggravates the inflammatory state in a vicious cycle that leads to the progression of COVID-19.
This evidence of the involvement of mitochondrial, platelets and dysbiosis in inflammatory processes and COVID opens up new avenues in the search for new therapeutic and preventive strategies in this serious disease and others that involve severe systemic inflammation.
BIBLIOGRAFÍA
(1) Saleh J, Peyssonnaux C, Singh KK, Edeas M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion. 2020;54:1-7. doi:10.1016/j.mito.2020.06.008
(2) Zhang, D., Guo, R., Lei, L., Liu, H., Wang, Y., Wang, Y., Dai, T., Zhang, T., Lai, Y., Wang, J., Liu, Z., He, A.O., Dwyer, M., Hu, J., 2020a. COVID-19 infection induces readily detectable morphological and inflammation-related phenotypic changes in peripheral blood monocytes, the severity of which correlate with patient outcome. medRxiv 2020.03.24.20042655.
(3) Zhu, H., Santo, A., Jia, Z., Li, Y., 2019. GPx4 in bacterial infection and polymicrobial sepsis: involvement of ferroptosis and pyroptosis. React. Oxyg. Species 7, 154–160.
(4) Aguirre, J.D., Culotta, V.C., 2012. Battles with iron: manganese in oxidative stress pro- tection. J. Biol. Chem. 287, 13541–13548.
(5) Wang, L., Wu, Q., Fan, Z., Xie, R., Wang, Z., Lu, Y., 2017. Platelet mitochondrial dys- function and the correlation with human diseases. Biochem. Soc. Trans. 45, 1213–1223.
(6) Melchinger, H., Jain, K., Tyagi, T., Hwa, J., 2019. Role of platelet mitochondria: life in a nucleus-free zone. Front. Cardiovasc. Med. 6, 1–11.
(7) Gou, W., Fu, Y., Yue, L., Chen, G., Cai, X., Shuai, M., Xu, F., Yi, X., Chen, H., Zhu, Y., Xiao, M., Jiang, Z., Miao, Z., Xiao, C., Shen, B., Wu, X., Zhao, H., Ling, W., Wang, J., Chen, Y., et al., 2020. Gut microbiota may underlie the predisposition of healthy individuals to COVID-19. medRix 1–44.
(8) Green, D., Galluzzi, L., Kroemer, G., 2011. Mitochondria and the autophagy- inflammation-cell death axis in organismal aging. Science (80-.) 333, 1109–1112
(9) Lin S-J, et al. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–16.
(11) Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013;15(Suppl 3):26–33.
(12) Lin SJ, et al. Life span extension by calorie restriction in S. cerevisiae requires NAD and SIR2. Science. 2000;289:2126–2128.